
蛋白质由氨基酸连接成肽链,在活细胞中承担大量关键功能。研究人员长期希望更精确地读取蛋白质的序列与结构信息,但与过去二十年快速发展的DNA测序相比,蛋白质测序工具在速度与规模上一直难以匹配。
斯坦福大学一支由生物工程师牵头的团队近日公布了一种新的蛋白质“可视化”与解码思路。相关研究于3月18日发表在《自然生物技术》上,提出一套化学流程,使研究人员能够利用现有快速、低成本的DNA测序平台来读取蛋白质序列信息。
该研究高级作者、斯坦福大学电气工程W·M·基克基金会教授、生物工程与放射学教授H·汤姆·索表示,自然界中蛋白质由DNA制造,而社会在过去二十年建立了能够快速且低成本测序大量DNA的技术体系;但蛋白质测序并未取得同等进展。索称,这项工作提出一种将蛋白质序列“转换回DNA序列”的技术路径,借此调用成熟的DNA测序能力。
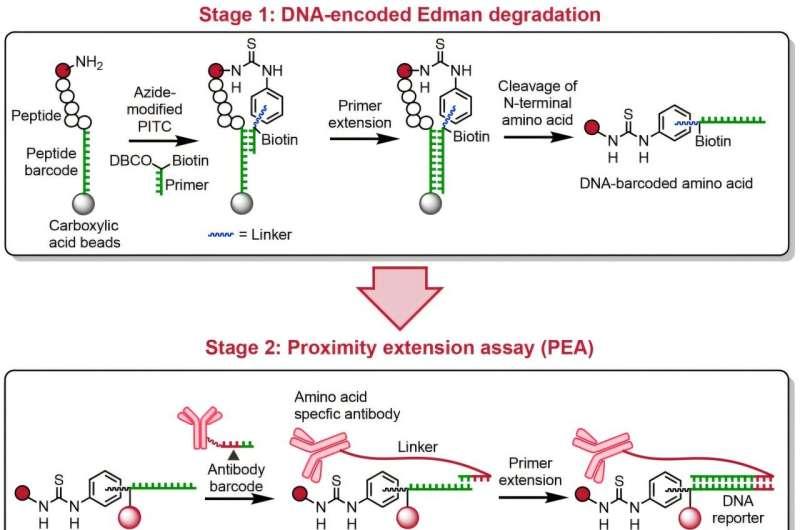
研究团队指出,蛋白质携带的生物信息并非DNA能够完全揭示。DNA更像构建细胞机器的说明书,而蛋白质是执行功能的“机器”,其折叠形成的复杂结构影响细胞生长、通讯、免疫应答以及疾病状态下的异常表现。由于缺乏与现代DNA测序速度相当的工具,蛋白质层面的精细读取一直是技术瓶颈。
研究人员解释,蛋白质测序难度较高的原因之一在于其由20种不同氨基酸构成,远多于DNA的四种碱基;同时,氨基酸的体积也更大,使得可靠检测与区分更具挑战。斯坦福研究工程师、论文第一作者郑立伟表示,与质谱等方法相比,新方法在同一样本中潜在可观察到的分子数量可显著提升:质谱往往在“射击”10亿到100亿个蛋白质分子后通常只能看到约一百万个分子,而该方法在潜在可见数量上可达到其1000倍,研究团队希望借此捕捉更多稀有蛋白质。
在技术路径上,团队提出的化学方法通过为每个肽分子引入DNA条形码来编码信息:利用抗体与循环条形码(研究中称为合成DNA)分别标记肽的身份并记录氨基酸在肽链中的位置。完成编码后,研究人员可使用常规DNA测序读取条形码,从而推断氨基酸的身份与位置。
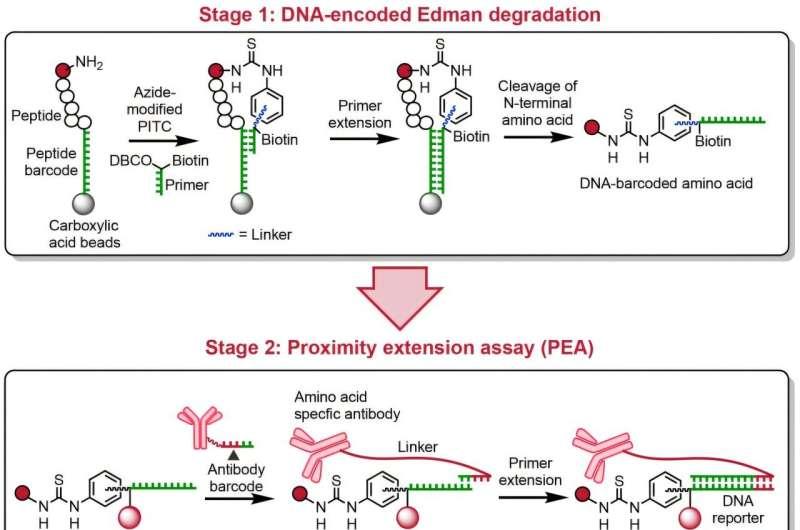
索表示,该方法的关键在于可在单分子水平对单个蛋白质进行测序,所需样本量很少,目标可达到单个细胞水平。研究团队认为,这种灵敏度可能支持更高分辨率的细胞多样性研究,用于观察看似相同的细胞在癌症等疾病情境下为何表现不同,并为在大样本中难以检测的稀有蛋白质研究提供新的可能。
研究团队以免疫疗法为例指出,CAR-T细胞疗法等通过改造患者免疫细胞识别并攻击癌细胞,已改变部分癌症治疗格局,但其为何仅对某些癌症类型和某些细胞类型有效仍存在未解问题。研究人员提出,可将对治疗有反应与无反应的免疫细胞分离,并进行单蛋白水平分析,以更清楚地比较差异并理解疗效差别。
研究还提到,该成果已引发商业化兴趣并完成许可,目标是将当前实验室流程转化为更易用的仪器化方案。索表示,团队希望最终实现类似现有DNA测序仪的使用体验,即研究人员“放入样本、按下按钮即可运行”。
研究团队同时强调,该方法仍处于早期阶段,距离广泛应用仍需进一步优化。不过,研究中提出的潜在规模——在数千个细胞中读取数十亿蛋白质分子——被认为是其区别于以往直接蛋白质测序尝试的重要特征。郑立伟表示,一旦将信息转换为DNA,就有机会利用DNA延长、复制等成熟机制来处理蛋白质序列信息。










