

研究团队提出一项面向QM/MM(量子力学/分子力学)模拟的新设计原则,核心在于利用电子态响应来客观、自动地确定量子力学(QM)计算区域,以解决多尺度分子模拟中QM与分子力学(MM)区域边界长期依赖经验划分的问题。
研究人员包括中央大学理工学部应用化学系森宏俊教授,以及御茶水女子大学一年级博士生小泽仁香和助理教授黑木奈穗子。相关成果以封面文章形式发表于《Advanced Science》。
QM/MM方法通过仅对化学相关区域进行量子力学处理、并以分子力学描述周围环境,被用于在可控计算成本下研究大型分子体系。然而,传统QM/MM模拟中QM与MM区域的边界往往由研究者凭经验或直觉设定,进而带来可重复性与预测可靠性等基础性问题。
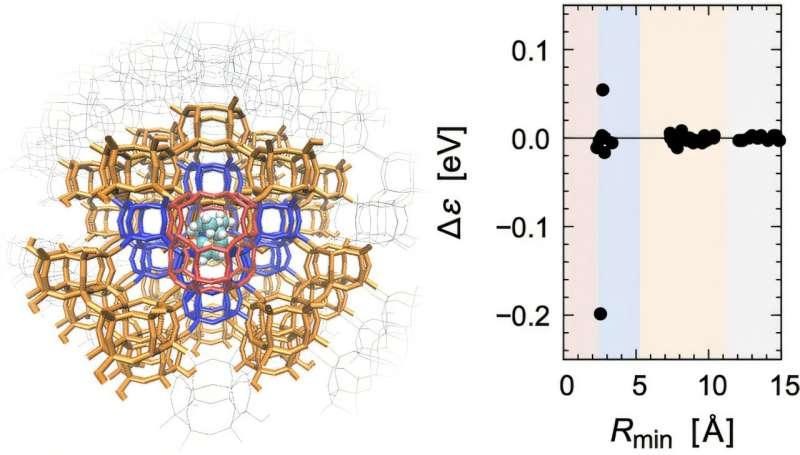
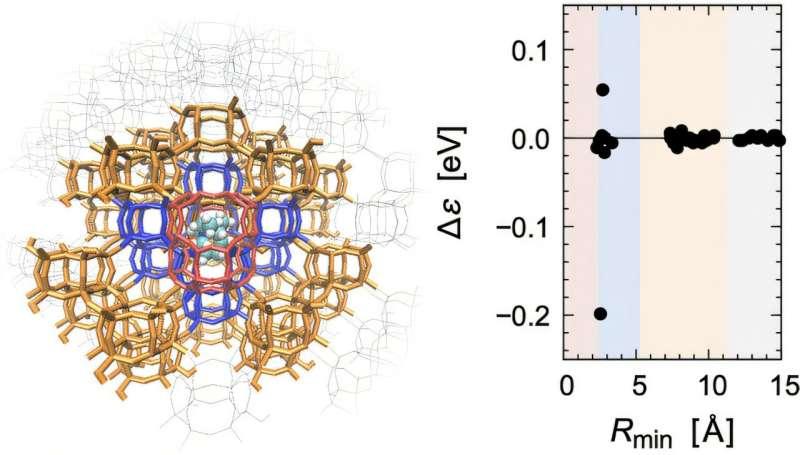
该研究将关注点放在化学反应与分子识别过程中出现的电子态响应,包括电荷重分布与分子轨道能量变化等。研究团队通过对整个体系开展基于片段的半经验分子轨道计算,并对电子态响应进行分析,提出一套具有物理依据的标准,用于识别应纳入量子力学处理的区域。
研究团队表示,这一原则使QM/MM边界的确定不再具有任意性,并可在不同体系与计算条件下保持一致适用性。
据介绍,该设计原则已在多类差异显著的体系中进行应用验证,包括无机多孔材料以及含抑制剂的生物分子体系。在相关案例中,能量评估均保持化学精度,研究团队据此认为所得QM/MM模型具备预测能力。该原则同时不依赖特定量子化学方法,可与更高层次的电子结构理论结合,例如密度泛函理论(DFT)与从头算方法。
研究团队指出,这项工作有助于推动QM/MM模拟从主要用于解释实验观察的工具,进一步发展为用于预测与设计分子功能及反应性的理论基础。
在后续工作方面,研究人员预计将该基于电子态响应的设计原则与机器学习和人工智能技术结合,并进一步用于由预测指导的目标实验验证,以期深化预测科学并推动复杂材料与反应体系的自动化设计。









