

研究人员在近期发表于《ACS Nano》的一项工作中提出,可基于纳米石墨烯分子单体在表面构筑一维聚合链,并通过化学手段调控电子与自旋结构,使同一分子骨架呈现两种不同的拓扑量子磁体平台。研究关注的核心在于:一维自旋链不仅可作为整体表现出量子特性,其关键性质还可能以边缘态形式出现在链的端点。
研究以TANGO型纳米石墨烯单体作为重复单元,设想将其连接成一维聚合物链。研究人员指出,该单体具备“电子可编程”特征:通过控制未配对电子在单体中的位置以及每个单体携带的未配对电子数量,可改变自旋出现方式及相邻单体之间的相互作用,从而在同一骨架上形成不同的耦合模式。
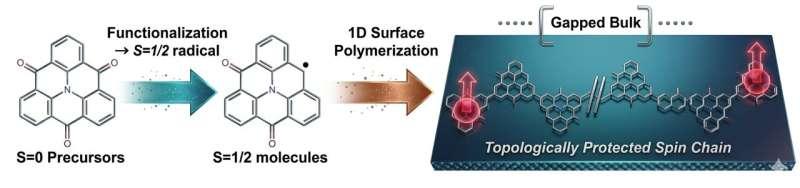
在该体系中,自旋来自未配对电子的量子磁矩。相邻自旋通过交换耦合相互作用:反铁磁耦合倾向于反向排列,铁磁耦合倾向于同向排列。研究人员强调,在分子体系里,化学结构会通过影响轨道重叠与局域电子结构而显著改变交换耦合强度与形式。
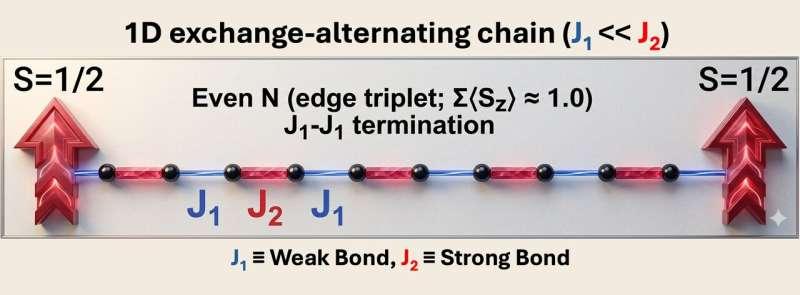
研究提出的第一类平台为“具有边缘磁矩的偶联S=1/2链”。在这一设定下,每个单体携带一个未配对电子,对应自旋1/2。链上相邻耦合呈强弱交替(偶联链),交替耦合会打开体态能隙,使激发需要一定能量;在特定终止方式下,链端会出现局域化的边缘磁矩。研究人员表示,这类边缘自旋并非外加,而是由强弱交替的耦合模式在边界处无法对称“闭合”所导致,并将其与Su–Schrieffer–Heeger(SSH)模型中的边界态类比。
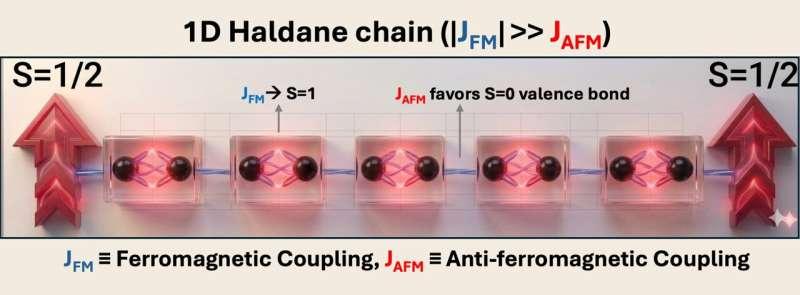
第二类平台为“Hund耦合的S=1 Haldane链”。在这一设定下,每个单体携带两个未配对电子,并倾向于在同一分子上自旋对齐(Hund规则),从而形成有效的自旋1位点。这些自旋1位点沿链以反铁磁方式耦合,构成Haldane链。研究人员指出,Haldane链的特征在于体态保持能隙,而开放端可承载分数化的自旋1/2边缘态,且相关边缘自由度由对称性保护。
为检验上述物理图景,研究团队采用两步方法:首先进行电子结构计算,以确定单体上未配对电子的分布并估算单体耦合后的主要交换相互作用;随后对得到的有效自旋模型开展适用于一维量子磁体的多体模拟。研究并未以复现所有微观细节为目标,而是聚焦于可用于识别拓扑相的稳健指标,包括有限的体态能隙、特征激发谱,以及随终止方式变化的边缘特征,并指出只要能隙保持打开,这些特征将持续存在。
研究人员在讨论潜在应用时对“量子比特”表述保持谨慎,强调并未宣称已实现可用量子器件。不过,其指出拓扑自旋链在端点自然产生局域且可寻址的自旋自由度,这类端点自由度是多种量子比特概念所依赖的关键要素。研究还提到两点实际吸引力:其一,边缘自旋在空间上局域化,便于在表面上通过扫描探针进行表征与操控;其二,边缘态与对称性保护的相位相关,适度缺陷不一定会立即消除相关特征。
研究人员表示,该工作强调了一条“化学优先”的拓扑量子物质设计路径:与在既有材料中寻找拓扑性质不同,该路线将拓扑作为设计目标,通过化学组装实现。下一步将是实验层面的制备与验证,包括在表面上构筑这些分子链、测量其磁性特征,并探测预测的边缘性质。
文章作者信息显示,麦吉尔大学物理系博士后研究员Khalid N. Anindya获得FRQNT博士后奖学金支持,其研究聚焦低维量子材料与基于分子的磁性,结合第一性原理电子结构方法、有效自旋模型及多体模拟,探索化学可编程平台以实现拓扑量子态与自旋量子比特相关概念。
本文源自Science X Dialog系列内容,研究人员在该系列中分享其已发表研究成果。









