

在航空航天、能源和计算领域的前沿企业不断寻求新材料以提升性能的背景下,理解材料在实际应用中的表现至关重要。然而,目前即使是最先进的模拟技术,也难以准确模拟大多数固体材料中复杂的化学结构,这导致材料创新过程耗时且成本高昂。
麻省理工学院的研究团队开发出一种能够准确模拟金属行为的新方法,无论其化学结构多么复杂。该方法核心是基于机器学习的模型,这些模型不仅加快了材料模拟速度,还提升了模拟的准确性。研究人员通过构建涵盖化学无序材料中多样化原子环境的训练数据集,显著优化了模型的表现。
在发表于《Science Advances》的一篇新论文中,团队展示了该方法能够在多种条件下准确预测多种金属合金的材料性能,并证明其在高成本实验条件下开发新材料的潜力。
麻省理工材料科学与工程系TDK职业发展教授Rodrigo Freitas表示:“本文聚焦于金属合金领域,但该方法同样适用于半导体等其他材料类型。它不局限于某一应用领域,能够助力开发可持续钢材、航空航天新材料等,这正是其令人兴奋之处。”
论文作者还包括博士生Killian Sheriff、Daniel Xiao、Yifan Cao以及谢菲尔德大学高级讲师Lewis R. Owen。
金属建模的挑战
材料性能主要由其内部化学元素的排列决定。即使化学成分相同,不同的原子排列也可能导致材料表现出脆性或可塑性。精确捕捉这种差异需要逐原子模拟材料,依赖于描述原子间相互作用的模型。过去二十年,机器学习成为构建此类模型的最准确方法。然而,现有模型在处理高度有序的化学结构时表现良好,但大多数固体材料的原子排列是无序且局部多变的,这给模型学习带来巨大挑战。
Freitas指出:“化学无序意味着存在大量不同的局部化学环境,这对机器学习模型来说非常难以学习。而我们实际使用的每一种金属都是化学无序的。”
问题的根源在于缺乏代表性的训练数据。现有生成训练数据的方法通常依赖大量计算资源,耗时超过10万小时,且难以适应材料成分的变化。
创新训练数据集构建
此前,Freitas团队开发了一种通过分析原子群的频率和间距来衡量材料化学复杂性的方法。本研究利用该技术,结合信息论方法,构建了更具代表性的训练数据集。该方法通过替换样本中的原子,减少重复,增加模型接触到的不同化学环境,从而提升训练数据的多样性和信息量。
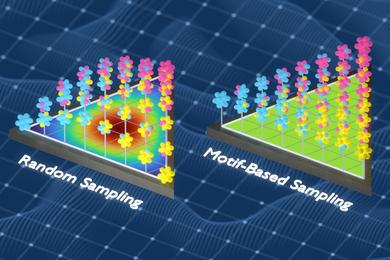
Freitas解释:“我们不断优化训练集,确保涵盖尽可能多的局部环境。重复的环境会被替换为模型未见过的新环境,使训练集更具信息量。”
基于这些数据集训练的模型,在材料性能预测上优于采用随机采样或其他流行采样方法训练的模型。
模型验证与应用
研究团队将该方法应用于一组化学成分多样的金属合金,结果显示其模型准确度超过了谷歌、微软等公司开发的更大规模模型。
Sheriff与Xiao、Cao合作,广泛测试了不同合金和性能的预测能力,并利用Owen的实验数据验证了模拟结果与实际合金中原子排序的吻合度。
实验室成果向工业应用转化
该方法能够捕捉样本数据中的“微妙能量偏好”,这些偏好决定了合金中相的形成、随温度和成分变化的演变,进而影响材料性能。Xiao领导的模拟显示,团队模型预测的相图与实验数据高度一致。相图是设计和加工合金的重要工具,帮助工程师了解不同条件下材料的稳定相。
Freitas表示:“相图是将材料建模与实际加工决策连接的关键。如果你在焊接、铸造或热处理合金,必须知道不同条件下可能形成的相。我们的目标是让这些预测既准确又易用,成为材料设计的常用工具。”
目前,团队正利用该方法研究合金成分变化对机械性能和抗辐射能力的影响,旨在设计出在恶劣环境下依然坚固耐损的材料。同时,他们致力于使该方法兼容现有材料工程师的工具和工作流程。
Freitas强调:“如果新方法不能融入工业现有操作流程,企业不会轻易改变。我们的目标是让预测结果在实际材料决策中发挥作用。”
本研究获得美国空军科学研究办公室的支持。









