

拜罗伊特大学研究人员近日公布一项利用人工智能加速液体性质计算的新方法,可用于预测化学势这一描述热力学平衡液体的关键物理量。相关研究成果已发表在《物理评论快报》(Physical Review Letters)。
研究团队指出,常见的人工智能建模路径多采用监督学习:以神经网络为代表的模型通过大量样本训练,直接输出某一特定目标量。此类方法在图像识别等任务中较为典型,即通过标注数据学习识别新图像中的目标对象。
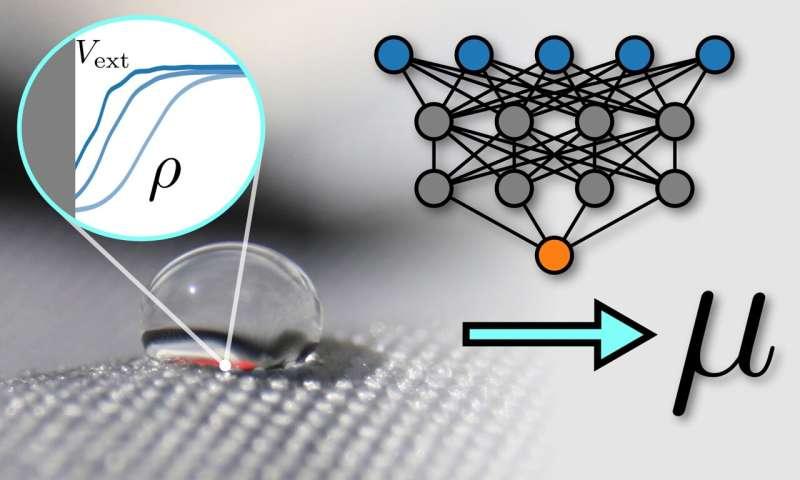
不过,研究人员表示,将“直接预测”的思路用于化学势并不容易。拜罗伊特大学理论物理学二系主任Matthias Schmidt教授称,化学势的确定通常依赖计算量巨大的算法,因此直接训练模型输出化学势面临挑战。为此,他与研究助理Florian Sammüller博士提出一种基于神经网络的新方案,将液体(更广义的软物质)的理论结构纳入模型设计,以实现对液体性质的高精度预测。
Schmidt介绍,该方法的核心在于模型并不直接学习化学势,而是学习“通用密度泛函”。这一密度泛函用于刻画液体内部的基本物理关系,并在不同系统之间保持不变。他以同一种液体覆盖在不同表面为例说明:即便表面结构或材料不同,液体仍遵循相同的基本物理规律;这些“内在”性质对应的正是机器学习所捕捉的通用密度泛函。

研究还指出,在学习得到的密度泛函与系统可观测性质(例如粒子密度分布和外部势)之间仍存在差异。该差异并非由模型补齐,而是由物理原理确定:基于热力学稳定性的一般考虑,这一剩余差异可唯一对应到化学势。
Sammüller表示,该方法将数据驱动学习与理论物理约束结合:人工智能给出通用框架,而化学势由既定物理条件导出,从而实现对化学势的间接且一致的确定,无需对化学势进行显式训练。他将其类比为图像识别任务中“模型在训练时未见过猫,却仍能识别猫”的情形。









