

药物分子设计常被研究人员形容为“分子俄罗斯方块”:化学家不断调整原子与结构片段的组合,直到获得具备潜力的候选分子。但在实践中,为了找到更优分子及其可行的合成路径,往往需要大量实验筛选,耗时且成本高。
据《自然》杂志近日发表的一项研究,研究人员利用机器学习建立了一套预测工作流程,旨在以更低成本评估反应组分的组合方式,并预测反应能否偏向生成目标“手性”产物,从而减少实验室反复试错。
该研究共同第一作者、犹他大学与加州大学洛杉矶分校联合博士后研究员Simone Gallarati表示,传统基于物理的计算化学工具可用于理解新反应,但若要对成千上万种潜在新分子进行预测,计算成本过高。团队希望训练一种既能对未测试反应做出较准确预测、又尽可能“廉价”的统计模型。
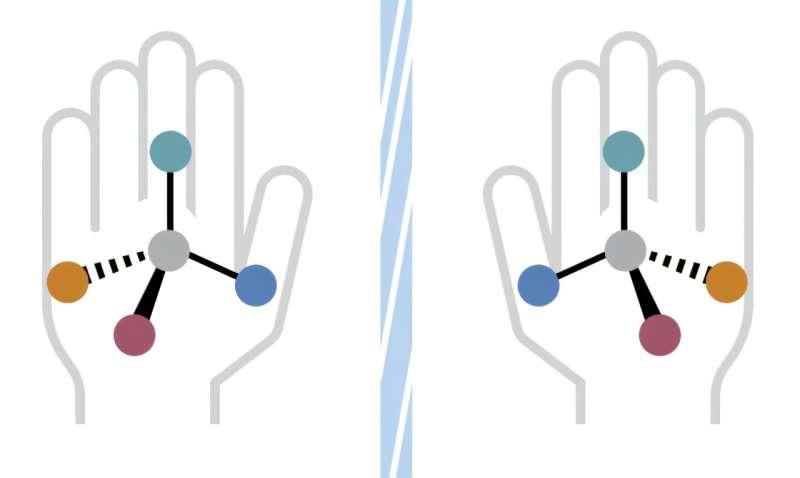
研究指出,许多分子存在镜像形式,即“手性”。左旋与右旋形式在药物中可能带来截然不同的效果,因此在合成中需要通过合适的催化剂、配体和底物组合,尽量选择性地产生目标手性版本。
在该工作中,研究团队将系统定位为一种“筛选器”:它可在计算层面评估大量化学结构,将反应组分转化为可供算法处理的数值信息,并据此预测不同组分组合对手性结果的影响。研究人员称,该模型所需输入较少,但能在一定范围内可靠预测组分行为,从而降低实验测试所需的时间、精力与费用。
犹他大学化学家、论文合著者Matthew Sigman表示,许多人工智能模型依赖大规模高质量数据集,而在化学领域获取此类数据通常昂贵且耗时。该研究的特点在于,允许用户用较小数据集建立模型,在准确预测已知反应的同时,将预测能力迁移到模型未见过的反应。
研究团队将方法聚焦于不对称交叉偶联反应。这类反应通过金属催化剂将两个碳基分子片段连接,构建更复杂的化合物;“不对称”则指反应被设计为偏向生成某一手性版本。论文称,在缺乏控制时,产物两种镜像形式可能接近50/50;而不对称反应可将目标形式比例提高至约95%,不需要的镜像约为5%。
研究介绍,不对称交叉偶联反应通常涉及金属、配体和底物等要素:金属催化剂促成碳基片段连接;配体与金属结合并影响反应发生的空间方向,从而决定产物三维构型,因而被视为控制手性的关键因素。
在模型训练方面,Gallarati及其团队选取了四篇关于不对称反应的学术论文作为训练数据,这些论文使用镍基催化剂并搭配不同配体,其中包括合著者Abigail Doyle与Sigman此前的研究。团队随后让系统预测训练数据未覆盖的假设组分反应结果,并逐步提高预测任务难度,使算法面对与原始训练数据差异更大的材料组合。
研究团队在Doyle实验室对部分预测结果进行了实验验证。该研究共同第一作者、加州大学洛杉矶分校博士生Erin Bucci表示,从实验化学家的角度看,该工具显著节省了实验时间:以往可能需要进行50至60个反应筛选,现在可缩减至5至10个,节省数周甚至数月;同时,由于每个反应组分都需要购买或自行合成,筛选次数减少也意味着材料开支下降。
论文作者称,尽管该工具在新型镍基反应体系中进行测试,但工作流程具备跨领域应用潜力。加州大学洛杉矶分校化学家、合著者Abigail Doyle表示,该方法并非“黑箱”,即使预测出现偏差,研究人员也能从中获得化学层面的信息,并结合专业判断进一步理解反应规律。
Sigman还指出,制药行业可能在反应优化与规模化生产场景中受益。例如,当企业需要为临床试验放大生产某化合物,并希望借鉴文献反应条件但该反应未在其目标化合物上验证时,这类预测工具可用于缩短优化周期、降低试错成本。









