

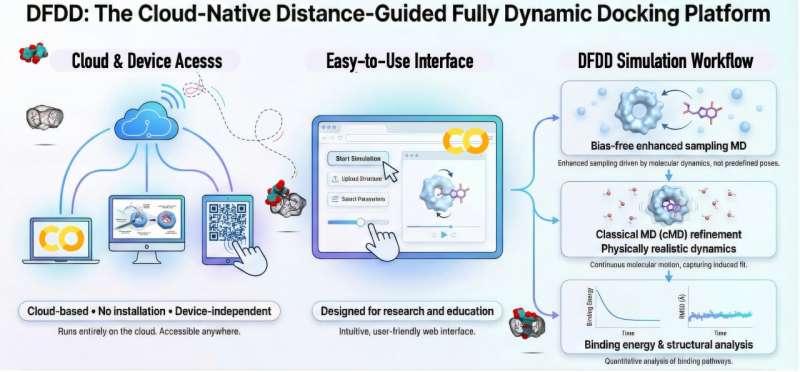
筑波大学计算科学中心研究人员开发了一套面向教学与研究的便捷模拟平台,旨在突破传统静态对接模拟的局限,为分子识别与超分子化学的教育培训及可重复研究提供新工具。该平台名为“距离引导全动态对接”(DFDD),定位为云端就绪的模拟框架,可用于直观展示分子结合的动态过程,并帮助理解客体—主客体晶体结构如何由分子相互作用逐步形成。
研究团队指出,分子如何结合是化学、生物学与药物设计中的关键问题,但现有不少工具主要提供静态结构“快照”,或对计算资源要求较高,超出许多研究人员与学生可承受范围。分子识别过程由连续运动驱动,分子柔性以及溶剂介导的相互作用在其中发挥重要作用。
在传统对接方法中,分子结合常借助静态晶体结构进行解释,且往往将分子视为刚性对象,导致多种结合路径难以被捕捉。尽管经典分子动力学(cMD)模拟能够描述连续过程,但其计算成本较高,且可重复性受限,影响了更广泛的应用。
相关研究发表在《化学信息与建模杂志》。论文展示了如何通过短时且可重复的模拟,在可访问的云平台(甚至平板设备)上捕捉真实的结合路径与类晶体结构,从而形成一个可用于学习、教学与研究的实用框架。
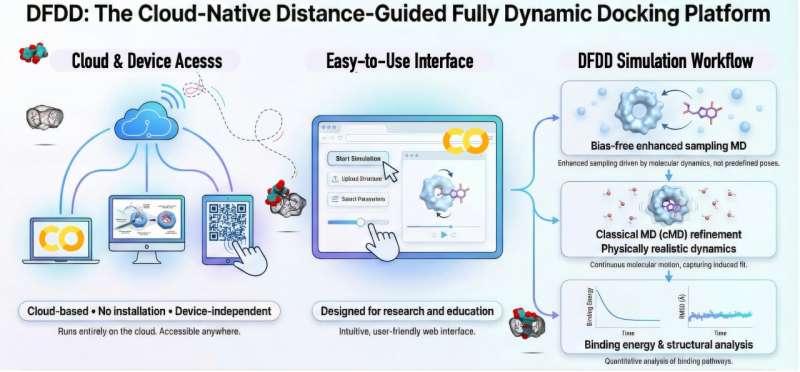
研究结果显示,DFDD能够可重复地恢复与实验确定晶体结构高度匹配的结合构型,同时揭示静态对接难以获得的瞬态构型。与从未结合状态启动的cMD模拟相比,DFDD在收敛速度、可重复性以及计算工作量方面表现更优。
研究团队表示,DFDD无需专用硬件或复杂的软件安装,面向广泛可用的云平台运行环境设计,例如Google Colab。通过在静态结构模型与全动态模拟之间建立衔接,该框架旨在降低动态对接的使用门槛。DFDD已在GitHub上免费提供。
发表评论
登录后才可评论。
去登录









